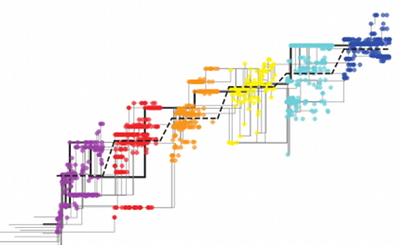
Human influenza A is one of the fastest-mutating viruses — and a laboratory for evolutionary innovation. But
how many of these mutations are adaptive and drive influenza's antigenic evolution? We analyze viral genome
sequences by a new method and find this number is surprisingly large. In a typical year, several driver
mutations coexist in different strains and compete for fixation. This is the first evidence of clonal
interference in a wild system.
Given that multiple clades compete for evolutionary success, which clade will win? We have developed a fitness
model for influenza that predicts the evolution of the viral population from one year to the next. Our analysis
provides a principled method to select influenza vaccine strains.
Concepts and Methods for Predicting Viral Evolution
Meijers, M., Ruchnewitz, D., Eberhardt, J., Karmakar, M., Luksza, M., Lässig,
M.,(2025)
in Influenza Virus. Methods in Molecular Biology, vol 2890. eds. Yamauchi, Y., Amorim, M.J., Humana,
New York, NY.
The seasonal human influenza virus
undergoes rapid evolution, leading to significant changes in circulating viral strains from year to year.
These changes are typically driven by adaptive mutations, particularly in the antigenic epitopes, the
regions of the viral surface protein hemagglutinin targeted by human antibodies. Here, we describe a
consistent set of methods for data-driven predictive analysis of viral evolution. Our pipeline integrates
four types of data: (1) sequence data of viral isolates collected on a worldwide scale, (2) epidemiological
data on incidences, (3) antigenic characterization of circulating viruses, and (4) intrinsic viral
phenotypes. From the combined analysis of these data, we obtain estimates of relative fitness for
circulating strains and predictions of clade frequencies for periods of up to 1 year. Furthermore, we obtain
comparative estimates of protection against future viral populations for candidate vaccine strains,
providing a basis for pre-emptive vaccine strain selection. Continuously updated predictions obtained from
the prediction pipeline for influenza and SARS-CoV-2 are available at
https://previr.app.
Nonequilibrium Antigen Recognition during Infections and Vaccinations
Morán-Tovar, R., Lässig,M., Phys.Rev.X 14, 031026, (2024)
The initial immune response to an
acute primary infection is a coupled process of antigen proliferation, molecular recognition by naive B cells,
and their subsequent clonal expansion. This process contains a fundamental problem: the recognition of an
exponentially time-dependent antigen signal. Here, we show that an efficient immune response must be
stringently constrained to B-cell lineages with high antigen binding affinity. We propose a tuned proofreading
mechanism for primary recognition of new antigens, where the molecular recognition machinery is adapted to the
complexity of the immune repertoire. We show that this process produces potent, specific, and fast recognition
of antigens, maintaining a spectrum of genetically distinct B-cell lineages as input for affinity maturation.
Our analysis maps the proliferation-recognition dynamics of a primary infection to a generalized
Luria-Delbrück process, akin to the dynamics of the classic fluctuation experiment. This map establishes a
link between signal recognition dynamics and evolution. We derive the resulting statistics of the activated
immune repertoire: Antigen binding affinity, expected size, and frequency of active B-cell clones are related
by power laws, which define the class of generalized Luria-Delbrück processes. Their exponents depend on the
antigen and B-cell proliferation rate, the number of proofreading steps, and the lineage density of the naive
repertoire. We extend the model to include spatiotemporal processes, including the diffusion-recognition
dynamics of a vaccination. Empirical data of activated mouse immune repertoires are found to be consistent
with activation involving about three proofreading steps. The model predicts key clinical characteristics of
acute infections and vaccinations, including the emergence of elite neutralizers and the effects of immune
aging. More broadly, our results establish infections and vaccinations as a new probe into the global
architecture and functional principles of immune repertoires.
Population immunity predicts evolutionary trajectories of SARS-CoV-2
Meijers M., Ruchnewitz D., Eberhardt J., Łuksza M., Lässig M., Cell 186,
5151–5164 (2023)
The large-scale evolution of the
SARS-CoV-2 virus has been marked by rapid turnover of genetic clades. New variants show intrinsic changes,
notably increased transmissibility, and antigenic changes that reduce cross-immunity induced by previous
infections or vaccinations. How this functional variation shapes global evolution has remained unclear. Here,
we establish a predictive fitness model for SARS-CoV-2 that integrates antigenic and intrinsic selection. The
model is informed by tracking of time-resolved sequence data, epidemiological records, and
cross-neutralization data of viral variants. Our inference shows that immune pressure, including contributions
of vaccinations and previous infections, has become the dominant force driving the recent evolution of
SARS-CoV-2. The fitness model can serve continued surveillance in two ways. First, it successfully predicts
the short-term evolution of circulating strains and flags emerging variants likely to displace the previously
predominant variant. Second, it predicts likely antigenic profiles of successful escape variants prior to
their emergence.
Steering and controlling evolution — from bioengineering to fighting pathogens
Lässig M., Mustonen V., Nourmohammad A., Nat Rev Genet, (2023)
Control interventions steer the
evolution of molecules, viruses, microorganisms or other cells towards a desired outcome. Applications range
from engineering biomolecules and synthetic organisms to drug, therapy and vaccine design against pathogens
and cancer. In all these instances, a control system alters the eco-evolutionary trajectory of a target
system, inducing new functions or suppressing escape evolution. Here, we synthesize the objectives, mechanisms
and dynamics of eco-evolutionary control in different biological systems. We discuss how the control system
learns and processes information about the target system by sensing or measuring, through adaptive evolution
or computational prediction of future trajectories. This information flow distinguishes pre-emptive control
strategies by humans from feedback control in biotic systems. We establish a cost–benefit calculus to gauge
and optimize control protocols, highlighting the fundamental link between predictability of evolution and
efficacy of pre-emptive control.
Stochasticity of infectious outbreaks and consequences for optimal interventions
Morán-Tovar R., Gruell H., Klein F., Lässig M., J. Phys. A: Math. Theor. 55
384008 (2022)
Global strategies to contain a
pandemic, such as social distancing and protective measures, are designed to reduce the overall transmission
rate between individuals. Despite such measures, essential institutions, including hospitals, schools, and
food producing plants, remain focal points of local outbreaks. Here we develop a model for the stochastic
infection dynamics that predicts the statistics of local outbreaks from observables of the underlying global
epidemics. Specifically, we predict two key outbreak characteristics: the probability of proliferation from a
first infection in the local community, and the establishment size, which is the threshold size of local
infection clusters where proliferation becomes likely. We derive these results using a contact network model
of communities, and we show how the proliferation probability depends on the contact degree of the first
infected individual. Based on this model, we suggest surveillance protocols by which individuals are tested
proportionally to their degree in the contact network. We characterize the efficacy of contact-based protocols
as a function of the epidemiological and the contact network parameters, and we show numerically that such
protocols outperform random testing.
Effective high-throughput RT-qPCR screening for SARS-CoV-2 infections in
children
Dewald F., Suárez I., Johnen R., Grossbach J., Moran-Tovar R., Steger G.,
Joachim
A., Rubio G.H., Fries M. , Behr F., Kley J., Lingnau A., Kretschmer A., Gude C., Baeza-Flores G., Laveaga
del
Valle D., Roblero-Hernandez A., Magana-Cerino J., Hernandez A.T., Ruiz-Quinones J., Schega K., Linne V.,
Junker L., Wunsch M., Heger E., Knops E., Di Cristanziano V., Meyer M., Hünseler C., Weber L.T., Lüers L.C.,
Quade G., Wisplinghoff H., Tiemann C., Zotz R., Jomaa H., Pranada A., Herzum I., Cullen P., Schmitz F.J.,
Philipsen P., Kirchner G., Knabbe C., Hellmich M., Buess M., Wolff A., Kossow A., Niessen J., Jeworutzki S.,
Schräpler J.-P., Lässig M., Dötsch J., Fätkenheuer D., Kaiser R., Beyer A., Rybniker J., Klein F., Nature
Communications, 13 (3640) , (2022)
Systematic SARS-CoV-2 testing is
a
valuable tool for infection control and surveillance. However, broad application of high sensitive RT-qPCR
testing in children is often hampered due to unpleasant sample collection, limited RT-qPCR capacities and
high
costs. Here, we developed a high-throughput approach (‘Lolli-Method’) for SARS-CoV-2 detection in children,
combining non-invasive sample collection with an RT-qPCR-pool testing strategy. SARS-CoV-2 infections were
diagnosed with sensitivities of 100% and 93.9% when viral loads were >106 copies/ml and >103 copies/ml in
corresponding Naso-/Oropharyngeal-swabs, respectively. For effective application of the Lolli-Method in
schools and daycare facilities, SEIR-modeling indicated a preferred frequency of two tests per week. The
developed test strategy was implemented in 3,700 schools and 698 daycare facilities in Germany, screening
over
800,000 individuals twice per week. In a period of 3 months, 6,364 pool-RT-qPCRs tested positive (0.64%),
ranging from 0.05% to 2.61% per week. Notably, infections correlated with local SARS-CoV-2 incidences and
with
a school social deprivation index. Moreover, in comparison with the alpha variant, statistical modeling
revealed a 36.8% increase for multiple (≥2 children) infections per class following infections with the
delta
variant. We conclude that the Lolli-Method is a powerful tool for SARS-CoV-2 surveillance and can support
infection control in schools and daycare facilities.
Clinical and Genomic Characterization of SARS CoV-2 infections in mRNA
Vaccinated Health Care Personnel in New York City
Robilotti E.V., Whiting K., Lucca A., Poon C., Guest R., McMillen T., Jani K.,
Solovyov A., Kelson S., Browne K., Freeswick S., Hohl T.M., Korenstein D., Ruchnewitz D., Lässig M., Łuksza
M., Greenbaum B., Seshan V.E., Babady N.E., Kamboj M., Clinical Infectious Diseases, ciab886, (2021)
Background
Vaccine-induced
clinical protection against SARS CoV-2 variants is an evolving target. There is limited genomic level data
on
SARS CoV-2 breakthrough infections and vaccine effectiveness (VE) since the global spread of the B.1.617.2
(Delta) variant.
Methods
In a retrospective study from November 1st, 2020 - August 31st , 2021,
divided as pre-Delta and Delta-dominant periods, laboratory-confirmed SARS CoV-2 infections among Healthcare
personnel (HCP) at a large tertiary cancer center in New York City (NYC) were examined to compare the weekly
infection rate-ratio in vaccinated, partially vaccinated, and unvaccinated HCP. We describe the clinical and
genomic epidemiologic features of post-vaccine infections to assess for selection of VOC/VOI in the early
post-vaccine period and impact of B.1.617.2 (Delta) variant domination on VE.
Results
Among 13,658
HCP in our cohort, 12,379 received at least one dose of an mRNA vaccine. In the pre-Delta period overall VE
was 94.5%. WGS of 369 isolates in the pre-Delta period did not reveal a clade bias for VOC/VOI specific to
post-vaccine infections. VE in the Delta dominant phase was 75.6%. No hospitalizations occurred among
vaccinated HCP in the entire study period, compared to 17 hospitalizations and one death among unvaccinated
HCP.
Conclusions
Findings show high VE among HCP in NYC in the pre-Delta phase, with moderate
decline in VE post-Delta emergence. SARS CoV-2 clades were similarly distributed among vaccinated and
unvaccinated infected HCP without apparent clustering during the pre-Delta period of diverse clade
circulation. Strong vaccine protection against hospitalization was maintained through the entire study
period.
Predicting in vivo escape dynamics of HIV-1 from a broadly neutralizing
antibody
Meijers M., Vanshylla K., Gruell H., Klein F., Lässig M., PNAS 118 (30)
e2104651118, (2021)
Broadly neutralizing antibodies
are
promising candidates for treatment and prevention of HIV-1 infections. Such antibodies can temporarily
suppress viral load in infected individuals; however, the virus often rebounds by escape mutants that have
evolved resistance. In this paper, we map a fitness model of HIV-1 interacting with broadly neutralizing
antibodies using in vivo data from a recent clinical trial. We identify two fitness factors, antibody dosage
and viral load, that determine viral reproduction rates reproducibly across different hosts. The model
successfully predicts the escape dynamics of HIV-1 in the course of an antibody treatment, including a
characteristic frequency turnover between sensitive and resistant strains. This turnover is governed by a
dosage-dependent fitness ranking, resulting from an evolutionary trade-off between antibody resistance and
its
collateral cost in drug-free growth. Our analysis suggests resistance–cost trade-off curves as a measure of
antibody performance in the presence of resistance evolution.
Antigenic waves of virus–immune coevolution
Marchi J., Lässig M., Walczak A.M., Mora T., PNAS 118 (27) e2103398118, (2021)
Viruses, such as influenza,
evolve
under the selection of host immune systems. Previously infected individuals become immune, forcing the virus
to find susceptible hosts or mutate, chasing it away in antigenic space. We formulate this viral escape
process in terms of a low-dimensional wave moving in antigenic space. The dimensionality of the antigenic
space impacts the persistence, as well as stability, of viral evolution. We uncover a characteristic
timescale
for the persistence of the viral strain, which is an order of magnitude longer than individual host immunity
and emerges collectively from the pressure of the chasing immune systems. These results offer intuition
about
the antigenic turnover of viruses and highlight the importance of the effective dimensionality of
coevolution.
Predicting evolution
Michael Lässig, Ville Mustonen, Aleksandra M. Walczak, Nature Ecology &
Evolution, (2017), doi:10.1038/s41559-017-0077
Abstract: The face of
evolutionary
biology is changing: from reconstructing and analysing the past to predicting future evolutionary pro-
cesses.
Recent developments include prediction of reproducible patterns in parallel evolution experiments,
forecasting
the future of individual populations using data from their past, and controlled manipulation of evolutionary
dynamics. Here we undertake a synthesis of central concepts for evolutionary predictions, based on examples
of
microbial and viral systems, can- cer cell populations, and immune receptor repertoires. These systems have
strikingly similar evolutionary dynamics driven by the competition of clades within a population. These
dynamics are the basis for models that predict the evolution of clade frequencies, as well as broad genetic
and phenotypic changes. Moreover, there are strong links between prediction and control, which are important
for interventions such as vaccine or therapy design. All of these are key elements of what may become a
predictive theory of evolution.
Epidemiological and evolutionary analysis of the 2014 Ebola virus outbreak
M. Łuksza, T.Bedford, and M. Lässig, arxiv:1411.1722 (2014)
Abstract: The 2014 epidemic of the
Ebola virus is governed by a genetically diverse viral population. In the early Sierra Leone outbreak, a
recent study has identified new mutations that generate genetically distinct sequence clades. Here we find
evidence that major Sierra Leone clades have systematic differences in growth rate and reproduction number.
If
this growth heterogeneity remains stable, it will generate major shifts in clade frequencies and influence
the
overall epidemic dynamics on time scales within the current outbreak. Our method is based on simple summary
statistics of clade growth, which can be inferred from genealogical trees with an underlying clade-specific
birth-death model of the infection dynamics. This method can be used to perform realtime tracking of an
evolving epidemic and identify emerging clades of epidemiological or evolutionary significance.
A predictive fitness model for influenza
M. Łuksza and M. Lässig, Nature, 507, 57-61 (2014)
The seasonal human influenza A
(H3N2)
virus undergoes rapid evolution, which produces significant year-to-year sequence turnover in the population
of circulating strains. Adaptive mutations respond to human immune challenge and occur primarily in
antigenic
epitopes, the antibody-binding domains of the viral surface protein haemagglutinin. Here we develop a
fitness
model for haemagglutinin that predicts the evolution of the viral population from one year to the next. Two
factors are shown to determine the fitness of a strain: adaptive epitope changes and deleterious mutations
outside the epitopes. We infer both fitness components for the strains circulating in a given year, using
population-genetic data of all previous strains. From fitness and frequency of each strain, we predict the
frequency of its descendent strains in the following year. This fitness model maps the adaptive history of
influenza A and suggests a principled method for vaccine selection. Our results call for a more
comprehensive
epidemiology of influenza and other fast-evolving pathogens that integrates antigenic phenotypes with other
viral functions coupled by genetic linkage.
Press releases
Press coverage
Clonal interference in the evolution of influenza
N. Strelkowa and M. Lässig, Genetics 192, 671 - 682 (2012)
Blah. The seasonal influenza A virus
undergoes rapid evolution to escape human immune response. Adaptive changes occur primarily in antigenic
epitopes, the antibody-binding domains of the viral haemagglutinin. This process involves recurrent
selective
sweeps, in which clusters of simultaneous nucleotide fixations in the haemagglutinin coding sequence are
observed about every 4 years. Here, we show that influenza A (H3N2) evolves by strong clonal interference.
This mode of evolution is a red queen race between viral strains with different beneficial mutations. Clonal
interference explains and quantifies the observed sweep pattern: We find an average of at least one strongly
beneficial amino acid substitution per year, and a given selective sweep has three to four driving mutations
on average. The inference of selection and clonal interference is based on frequency time-series of
single-nucleotide polymorphisms, which are obtained from a sample of influenza genome sequences over 39
years.
Our results imply that mode and speed of influenza evolution are governed not only by positive selection
within, but also by background selection outside antigenic epitopes: immune adaptation and conservation of
other viral functions interfere with each other. Hence, adapting viral proteins are predicted to be
particularly brittle. We conclude that a quantitative understanding of influenza's evolutionary and
epidemiological dynamics must be based on all genomic domains and functions coupled by clonal interference.
go back