Can the complexity of molecular interaction networks account for the bewildering diversity of organisms and species, and for their rapid adaptation to different environments? Interactions between genes are themselves encoded in the genome: Specific proteins (called transcription factors) bind to their corresponding target sites on the DNA, thereby enhancing or reducing the transcription of a nearby gene. Our work on regulation is mainly concerned with fitness effects of regulatory interactions, their inference from genomic data, and their consequences for the evolution of regulation. This work links the biophysics of protein-DNA interactions with the evolution of their biological function. In a recent experimental study, we carry this approach to the fitness landscape of an entire metabolic pathway, which depends on its regulatory sequence and on the environment of the cell. Another recent paper explores the joint fitness effects of nucleosome positioning and regulatory binding sites in the yeast genome.
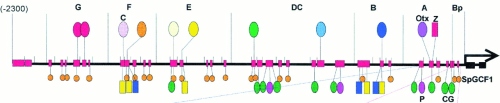
A segment of regulatory DNA in a eukaryotic cell with many binding sites for various transcription factors (Davidson lab).
Fitness landscape of nucleosome positioning
D. Weghorn and M. Lässig, Proc. Natl. Acad. Sci. 110, 10988–93 (2013)
Histone–DNA complexes, so-called nucleosomes, are the building blocks of DNA packaging in eukaryotic cells. The histone-binding affinity of a local DNA segment depends on its elastic properties and determines its accessibility within the nucleus, which plays an important role in the regulation of gene expression. Here, we derive a fitness landscape for intergenic DNA segments in yeast as a function of two molecular phenotypes: their elasticity-dependent histone affinity and their coverage with transcription factor binding sites. This landscape reveals substantial selection against nucleosome formation over a wide range of both phenotypes. We use it as the core component of a quantitative evolutionary model for intergenic DNA segments. This model consistently predicts the observed diversity of histone affinities within wild Saccharomyces paradoxus populations, as well as the affinity divergence between neighboring Saccharomyces species. Our analysis establishes histone binding and transcription factor binding as two separable modes of sequence evolution, each of which is a direct target of natural selection.
Formation of regulatory modules by local sequence duplication
A. Nourmohammad and M. Lässig, PLoS Comp. Biol., PLoS Comput Biol 7, e1002167 (12 pages), (2011)
Turnover of regulatory sequence and function is an important part of molecular evolution. But what are the modes of sequence evolution leading to rapid formation and loss of regulatory sites? Here, we show that a large fraction of neighboring transcription factor binding sites in the fly genome have formed from a common sequence origin by local duplications. This mode of evolution is found to produce regulatory information: duplications can seed new sites in the neighborhood of existing sites. Duplicate seeds evolve subsequently by point mutations, often towards binding a different factor than their ancestral neighbor sites. These results are based on a statistical analysis of 346 cis-regulatory modules in the Drosophila melanogaster genome, and a comparison set of intergenic regulatory sequences in Saccharomyces cerevisiae. In fly regulatory modules, pairs of binding sites show significantly enhanced sequence similarity up to distances of about 50 bp. We analyze these data in terms of an evolutionary model with two distinct modes of site formation: (i) evolution from independent sequence origin and (ii) divergent evolution following duplication of a common ancestor sequence. Our results suggest that pervasive formation of binding sites by local sequence duplications distinguishes the complex regulatory architecture of higher eukaryotes from the simpler architecture of unicellular organisms.
Nonlinear fitness landscape of a molecular pathway
Lilia Perfeito, Stéphane Ghozzi, Johannes Berg, Karin Schnetz, Michael Lässig, PloS Genetics 7, e1002160 (10 pages), (2011)
Genes are regulated because their expression involves a fitness cost to the organism. The production of proteins by transcription and translation is a well-known cost factor, but the enzymatic activity of the proteins produced can also reduce fitness, depending on the internal state and the environment of the cell. Here, we map the fitness costs of a key metabolic network, the lactose utilization pathway in Escherichia coli. We measure the growth of several regulatory lac operon mutants in different environments inducing expression of the genes. We find a strikingly nonlinear fitness landscape, which depends on the production rate and on the activity rate of the lac proteins. A simple fitness model of the lac pathway, based on elementary biophysical processes, predicts the growth rate of all observed strains. The nonlinearity of fitness is explained by a feedback loop: production and activity of the lac proteins reduce growth, but growth also affects the density of these molecules. This nonlinearity has important consequences for molecular function and evolution. It generates a cliff in the fitness landscape, beyond which populations cannot maintain growth. In viable populations, there is an expression barrier of the lac genes, which cannot be exceeded in any stationary growth process. Furthermore, the nonlinearity determines how the fitness of operon mutants depends on the inducer environment. We argue that fitness nonlinearities, expression barriers, and gene-environment interactions are generic features of fitness landscapes for metabolic pathways, and we discuss their implications for the evolution of regulation.
Energy-dependent fitness: a quantitative model for the evolution of yeast transcription factor binding sites
V. Mustonen, J. Kinney, CG. Callan Jr, and M. Lässig, Proc. Natl. Acad. Sci. 105, 12376-81, (2008)
We present a genomewide cross-species analysis of regulation for broad-acting transcription factors in yeast. Our model for binding site evolution is founded on biophysics: the binding energy between transcription factor and site is a quantitative phenotype of regulatory function, and selection is given by a fitness landscape that depends on this phenotype. The model quantifies conservation, as well as loss and gain, of functional binding sites in a coherent way. Its predictions are supported by direct cross-species comparison between four yeast species. We find ubiquitous compensatory mutations within functional sites, such that the energy phenotype and the function of a site evolve in a significantly more constrained way than does its sequence. We also find evidence for substantial evolution of regulatory function involving point mutations as well as sequence insertions and deletions within binding sites. Genes lose their regulatory link to a given transcription factor at a rate similar to the neutral point mutation rate, from which we infer a moderate average fitness advantage of functional over nonfunctional sites. In a wider context, this study provides an example of inference of selection acting on a quantitative molecular trait.
From biophysics to evolutionary genetics: statistical aspects of gene regulation
M. Lässig, BMC Bioinformatics 8 Suppl 6, S7, (2007)
This is an introductory review on how genes interact to produce biological functions. Transcriptional interactions involve the binding of proteins to regulatory DNA. Specific binding sites can be identified by genomic analysis, and these undergo a stochastic evolution process governed by selection, mutations, and genetic drift. We focus on the links between the biophysical function and the evolution of regulatory elements. In particular, we infer fitness landscapes of binding sites from genomic data, leading to a quantitative evolutionary picture of regulation.
Evolutionary population genetics of promoters: predicting binding sites and functional phylogenies
V. Mustonen and M. Lässig, Proc. Natl. Acad. Sci. 103, 10967, (2005)
We study the evolution of transcription factor-binding sites in prokaryotes, using an empirically grounded model with point mutations and genetic drift. Selection acts on the site sequence via its binding affinity to the corresponding transcription factor. Calibrating the model with populations of functional binding sites, we verify this form of selection and show that typical sites are under substantial selection pressure for functionality: for cAMP response protein sites in Escherichia coli, the product of fitness difference and effective population size takes values 2NΔF of order 10. We apply this model to cross-species comparisons of binding sites in bacteria and obtain a prediction method for binding sites that uses evolutionary information in a quantitative way. At the same time, this method predicts the functional histories of orthologous sites in a phylogeny, evaluating the likelihood for conservation or loss or gain of function during evolution. We have performed, as an example, a cross-species analysis of
E. coli,
Salmonella typhimurium, and
Yersinia pseudotuberculosis. Detailed lists of predicted sites and their functional phylogenies are available.
Adaptive evolution of transcription factor binding sites
J. Berg, S. Willmann, and M. Lässig, BMC Evol. Biol. 4, 42, (2004)
Background
The regulation of a gene depends on the binding of transcription factors to specific sites located in the regulatory region of the gene. The generation of these binding sites and of cooperativity between them are essential building blocks in the evolution of complex regulatory networks. We study a theoretical model for the sequence evolution of binding sites by point mutations. The approach is based on biophysical models for the binding of transcription factors to DNA. Hence we derive empirically grounded fitness landscapes, which enter a population genetics model including mutations, genetic drift, and selection.
Results
We show that the selection for factor binding generically leads to specific correlations between nucleotide frequencies at different positions of a binding site. We demonstrate the possibility of rapid adaptive evolution generating a new binding site for a given transcription factor by point mutations. The evolutionary time required is estimated in terms of the neutral (background) mutation rate, the selection coefficient, and the effective population size.
Conclusions
The efficiency of binding site formation is seen to depend on two joint conditions: the binding site motif must be short enough and the promoter region must be long enough. These constraints on promoter architecture are indeed seen in eukaryotic systems. Furthermore, we analyse the adaptive evolution of genetic switches and of signal integration through binding cooperativity between different sites. Experimental tests of this picture involving the statistics of polymorphisms and phylogenies of sites are discussed.
Stochastic evolution of transcription factor binding sites
J. Berg and M. Lässig, Biophysics (Moscow) 48, Suppl. 1 (2003)
A key step in the process of genetic transcription is the binding of one or several transcription factors to specific sites in the regulatory region of a gene. These binding sites may differ strongly across even closely related species, and the generation of new binding sites is an essential part of the evolution of regulatory networks. In this paper we consider the sequence evolution of binding sites, using empirically grounded fitness landscapes. We demonstrate how a new binding site for a given transcription factor may be generated de novo, and estimate the time required for this process in terms of the neutral mutation rate, the selection coefficient, and the effective population size. We also consider how several sites binding to the same type of factor can coexist in the regulatory region of a gene.
go back 