Adaptive evolution and dynamical fitness models
Evolution is a quest for innovation. Organisms adapt to changing natural selection by evolving new phenotypes.
Can we read this dynamics in their genomes? We have developed a nonequilibrium theory of molecular evolution,
which is based on explicitly time-dependent models of selection and adaptive genome evolution in
response. Our approach extends the static concept of fitness landscapes to dynamic fitness seascapes. It leads to
new tests for adaptive evolution in genomic data. Recently, we extended this approach to adaptive processes of
asexual populations, which take place under genetic linkage.
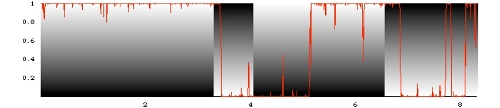
Stochastic evolution of a genomic locus under
mutations, reproductive fluctuations (genetic drift), and time-dependent selection. The graph shows the
time-dependent fraction of individuals carrying one of the two alleles.
Towards evolutionary predictions: Current promises and challenges
Wortel M., Agashe D., Bailey S., Bank C., Bisschop K., Blankers T., Cairns J.,
Colizzi E., Cusseddu D.,
Desai M., van Dijk B., Egas M., Ellers J., Groot A., Heckel D., Johnson M., Kraaijeveld K., Krug J., Laan L.,
Lässig M., Lind P., Meijer J., Noble L., Okasha S., Rainey P., Rozen D., Shitut S., Tans S., Tenaillon O.,
Teotónio H., de Visser J.,
Visser M., Vroomans R., Werner G., Wertheim B., Pennings P., Evolutionary Applications, 3-21, 16(1), (2023)
Evolution has traditionally been a
historical
and descriptive science, and predicting future evolutionary processes has long been considered impossible.
However, evolutionary predictions are increasingly being developed and used in medicine, agriculture,
biotechnology and
conservation biology. Evolutionary predictions may be used for different purposes, such as to prepare for the
future,
to try and change the course of evolution or to determine how well we understand evolutionary processes.
Similarly, the exact aspect of the evolved population that we want to predict may also differ.
For example, we could try to predict which genotype will dominate, the fitness of the population or the
extinction
probability of a population. In addition, there are many uses of evolutionary predictions that may not always
be recognized
as such. The main goal of this review is to increase awareness of methods and data in different research
fields by showing
the breadth of situations in which evolutionary predictions are made. We describe how diverse evolutionary
predictions share
a common structure described by the predictive scope, time scale and precision. Then, by using examples
ranging from SARS-CoV2
and influenza to CRISPR-based gene drives and sustainable product formation in biotechnology, we discuss the
methods for predicting evolution, the factors that affect predictability and how predictions can be used to
prevent evolution in undesirable directions or to promote beneficial evolution (i.e. evolutionary control). We
hope that this review will stimulate collaboration between fields by establishing a common language for
evolutionary predictions.
Two modes of evolution shape bacterial strain diversity in the mammalian gut for
thousands of generations
Frazão N., Konrad A., Amicone M., Seixas E., Güleresi D., Lässig M. and Gordo I.,
Nature Communications, 13 (5604) , (2022)
How and at what pace bacteria
evolve when colonizing healthy hosts remains unclear. Here, by monitoring evolution for more than six thousand
generations in the mouse gut, we show that the successful colonization of an invader Escherichia coli depends
on the diversity of the existing microbiota and the presence of a closely related strain. Following
colonization, two modes of evolution were observed: one in which diversifying selection leads to long-term
coexistence of ecotypes and a second in which directional selection propels selective sweeps. These modes can
be quantitatively distinguished by the statistics of mutation trajectories. In our experiments, diversifying
selection was marked by the emergence of metabolic mutations, and directional selection by acquisition of
prophages, which bring their own benefits and costs. In both modes, we observed parallel evolution, with
mutation accumulation rates comparable to those typically observed in vitro on similar time scales. Our
results show how rapid ecotype formation and phage domestication can be in the mammalian gut.
Metabolic fitness landscapes predict the evolution of antibiotic resistance
Pinheiro F., Warsi O., Andersson D.I., Lässig M., Nature Ecol. Evol, in press
(2021)
Bacteria evolve resistance to
antibiotics by a multitude of mechanisms. A central, yet unsolved question is how resistance evolution affects
cell growth at different drug levels. Here, we develop a fitness model that predicts growth rates of common
resistance mutants from their effects on cell metabolism. The model maps metabolic effects of resistance
mutations in drug-free environments and under drug challenge; the resulting fitness trade-off defines a Pareto
surface of resistance evolution. We predict evolutionary trajectories of growth rates and resistance levels,
which characterize Pareto resistance mutations emerging at different drug dosages. We also predict the
prevalent resistance mechanism depending on drug and nutrient levels: low-dosage drug defence is mounted by
regulation, evolution of distinct metabolic sectors sets in at successive threshold dosages. Evolutionary
resistance mechanisms include membrane permeability changes and drug target mutations. These predictions are
confirmed by empirical growth inhibition curves and genomic data of Escherichia coli populations. Our results
show that resistance evolution, by coupling major metabolic pathways, is strongly intertwined with systems
biology and ecology of microbial populations.
Adaptive evolution of hybrid bacteria by horizontal gene transfer
Power J.J., Pinheiro F., Pompei S., Kovacova V., Yüksel M., Rathmann I., Förster
M., Lässig M., Maier B., PNAS 11, 118 (10), (2021)
In a parallel evolution experiment,
we probe lateral gene transfer between two Bacillus subtilis lineages close to the species boundary. We show
that laboratory evolution by horizontal gene transfer can rapidly generate hybrid organisms with broad genomic
and functional alterations. By combining genomics, transcriptomics, fitness assays, and statistical modeling,
we map the selective effects underlying gene transfer. We show that transfer takes place under genome-wide
positive and negative selection, generating a net fitness increase in hybrids. The evolutionary dynamics
efficiently navigates this fitness landscape, finding viable paths with increasing fraction of transferred
genes.
Adaptive Evolution of Gene Expression in Drosophila
Armita Nourmohammad, Joachim Rambeau, Torsten Held, Viera Kovacova, Johannes
Berg, Michael Lässig, Cell Reports 20, 1385-1395 (2017)
Gene expression levels are
important quantitative traits that link genotypes to molecular functions and fitness. In Drosophila,
population-genetic studies have revealed substantial adaptive evolution at the genomic level, but the
evolutionary modes of gene expression remain controversial. Here, we present evidence that adaptation
dominates the evolution of gene expression levels in flies. We show that 64% of the observed expression
divergence across seven Drosophila species are adaptive changes driven by directional selection. Our results
are derived from time-resolved data of gene expression divergence across a family of related species, using a
probabilistic inference method for gene-specific selection. Adaptive gene expression is stronger in specific
functional classes, including regulation, sensory perception, sexual behavior, and morphology. Moreover, we
identify a large group of genes with sex-specific adaptation of expression, which predominantly occurs in
males. Our analysis opens an avenue to map system-wide selection on molecular quantitative traits
independently of their genetic basis.
Predicting evolution
Michael Lässig, Ville Mustonen, Aleksandra M. Walczak, Nature Ecol. Evol. 1, 077
(9 pages) (2017)
Abstract: The face of evolutionary
biology is changing: from reconstructing and analysing the past to predicting future evolutionary pro- cesses.
Recent developments include prediction of reproducible patterns in parallel evolution experiments, forecasting
the future of individual populations using data from their past, and controlled manipulation of evolutionary
dynamics. Here we undertake a synthesis of central concepts for evolutionary predictions, based on examples of
microbial and viral systems, can- cer cell populations, and immune receptor repertoires. These systems have
strikingly similar evolutionary dynamics driven by the competition of clades within a population. These
dynamics are the basis for models that predict the evolution of clade frequencies, as well as broad genetic
and phenotypic changes. Moreover, there are strong links between prediction and control, which are important
for interventions such as vaccine or therapy design. All of these are key elements of what may become a
predictive theory of evolution.
Adaptive evolution of molecular phenotypes
T. Held, A. Nourmohammad, and M. Lässig, J. Stat. Mech., in press (2014)
Molecular phenotypes link genomic
information with organismic functions, fitness, and evolution. Quantitative traits are complex phenotypes that
depend on multiple genomic loci. In this paper, we study the adaptive evolution of a quantitative trait under
time-dependent selection, which arises from environmental changes or through fitness interactions with other
co-evolving phenotypes. We analyze a model of trait evolution under mutations and genetic drift in a
single-peak fitness seascape. The fitness peak performs a constrained random walk in the trait amplitude,
which determines the time-dependent trait optimum in a given population. We derive analytical expressions for
the distribution of the time-dependent trait divergence between populations and of the trait diversity within
populations. Based on this solution, we develop a method to infer adaptive evolution of quantitative traits.
Specifically, we show that the ratio of the average trait divergence and the diversity is a universal function
of evolutionary time, which predicts the stabilizing strength and the driving rate of the fitness seascape.
From an information-theoretic point of view, this function measures the macro-evolutionary entropy in a
population ensemble, which determines the predictability of the evolutionary process. Our solution also
quantifies two key characteristics of adapting populations: the cumulative fitness flux, which measures the
total amount of adaptation, and the adaptive load, which is the fitness cost due to a population's lag behind
the fitness peak.
Universality and predictability in molecular quantitative genetics
A. Nourmohammad*, T. Held*, and M. Lässig, Current Opinion in Genetics and
Development 23, 684-93 (2013)
(*) equal contributions
Molecular traits, such as gene
expression levels or protein binding affinities, are increasingly accessible to quantitative measurement by
modern high-throughput techniques. Such traits measure molecular functions and, from an evolutionary point of
view, are important as targets of natural selection. We review recent developments in evolutionary theory and
experiments that are expected to become building blocks of a quantitative genetics of molecular traits. We
focus on universal evolutionary characteristics: these are largely independent of a trait's genetic basis,
which is often at least partially unknown. We show that universal measurements can be used to infer selection
on a quantitative trait, which determines its evolutionary mode of conservation or adaptation. Furthermore,
universality is closely linked to predictability of trait evolution across lineages. We argue that universal
trait statistics extends over a range of cellular scales and opens new avenues of quantitative evolutionary
systems biology.
Chance and risk in adaptive evolution
M.Lässig, Proc. Natl. Acad. Sci. 109, 4719-20, (2012)
Emergent Neutrality in Adaptive Asexual Evolution
Stephan Schiffels, Gergely Szöllösi, Ville Mustonen, and Michael Lässig, Genetics
189, 1361 - 75, (2011)
In non-recombining genomes, genetic
linkage can be an important evolutionary force. Linkage generates interference interactions, by which
simultaneously occurring mutations affect each other’s chance of fixation. Here, we develop a comprehensive
model of adaptive evolution in linked genomes. By an approximate analytical solution, we predict fixation
rates of beneficial and deleterious mutations, as well as the statistics of beneficial and deleterious alleles
at fixed genomic sites. We find that interference interactions generate a regime of effective neutrality: all
genomic sites with selection coefficients smaller in magnitude than a characteristic threshold have nearly
random fixed alleles, and both beneficial and deleterious mutations at these sites have nearly neutral
fixation rates. We show that this dynamics limits not only the speed of adaptation, but also a population’s
degree of adaptation in its current environment. Our model integrates interference between beneficial
mutations, genetic hitchhiking of weakly selected mutations, and background selection by strongly deleterious
mutations into a unified framework of interference interactions. We apply the model to different adaptive
scenarios: stationary adaptation in a time-dependent environment, and approach to equilibrium in a fixed
environment (as in long-term evolution experiments). In both cases, the analytical predictions are in good
agreement with numerical simulations. Our results suggest that interference can severely compromise biological
functions in an adapting population, which sets viability limits on adaptive evolution under linkage.
Fitness flux and ubiquity of adaptive evolution
V. Mustonen and M. Lässig, Proc. Natl. Acad. Sci. 107, 4248-53, (2010)
Natural selection favors fitter
variants in a population, but actual evolutionary processes may decrease fitness by mutations and genetic
drift. How is the stochastic evolution of molecular biological systems shaped by natural selection? Here, we
derive a theorem on the fitness flux in a population, defined as the selective effect of its genotype
frequency changes. The fitness-flux theorem generalizes Fisher's fundamental theorem of natural selection to
evolutionary processes including mutations, genetic drift, and time-dependent selection. It shows that a
generic state of populations is adaptive evolution: there is a positive fitness flux resulting from a surplus
of beneficial over deleterious changes. In particular, stationary nonequilibrium evolution processes are
predicted to be adaptive. Under specific nonstationary conditions, notably during a decrease in population
size, the average fitness flux can become negative. We show that these predictions are in accordance with
experiments in bacteria and bacteriophages and with genomic data in Drosophila. Our analysis establishes
fitness flux as a universal measure of adaptation in molecular evolution.
From fitness landscapes to seascapes: non-equilibrium dynamics of selection and
adaptation
V. Mustonen and M. Lässig, Trends Genet 25, 111-9, (2009)
Evolution is a quest for innovation.
Organisms adapt to changing natural selection by evolving new phenotypes. Can we read this dynamics in their
genomes? Not every mutation under positive selection responds to a change in selection: beneficial changes
also occur at evolutionary equilibrium, repairing previous deleterious changes and restoring existing
functions. Adaptation, by contrast, is viewed here as a non-equilibrium phenomenon: the genomic response to
time-dependent selection. Our approach extends the static concept of fitness landscapes to dynamic fitness
seascapes. It shows that adaptation requires a surplus of beneficial substitutions over deleterious ones.
Here, we focus on the evolution of yeast and Drosophila genomes, providing examples where adaptive evolution
can and cannot be inferred, despite the presence of positive selection
Molecular evolution under fitness fluctuations
V. Mustonen and M. Lässig, Phys Rev Lett. 100, 108101, (2008)
Molecular evolution is a stochastic
process governed by fitness, mutations, and reproductive fluctuations in a population. Here, we study
evolution where fitness itself is stochastic, with random switches in the direction of selection at individual
genomic loci. As the correlation time of these fluctuations becomes larger than the diffusion time of
mutations within the population, fitness changes from an annealed to a quenched random variable. We show that
the rate of evolution has its maximum in the crossover regime, where both time scales are comparable. Adaptive
evolution emerges in the quenched fitness regime (evidence for such fitness fluctuations has recently been
found in genomic data). The joint statistical theory of reproductive and fitness fluctuations establishes a
conceptual connection between evolutionary genetics and statistical physics of disordered system
Adaptations to fluctuating selection in Drosophila
V. Mustonen and M. Lässig, Proc. Natl. Acad. Sci. 104, 2277-82, (2007)
Time-dependent selection causes the
adaptive evolution of new phenotypes, and this dynamics can be traced in genomic data. We have analyzed
polymorphisms and substitutions in Drosophila, using a more sensitive inference method for adaptations than
the standard population-genetic tests. We find evidence that selection itself is strongly time-dependent, with
changes occurring at nearly the rate of neutral evolution. At the same time, higher than previously estimated
levels of selection make adaptive responses by a factor 10-100 faster than the pace of selection changes,
ensuring that adaptations are an efficient mode of evolution under time-dependent selection. The rate of
selection changes is faster in noncoding DNA, i.e., the inference of functional elements can less be based on
sequence conservation than for proteins. Our results suggest that selection acts not only as a constraint but
as a major driving force of genomic change.
go back 