Recent projects
Structure and evolution of regulatory DNA
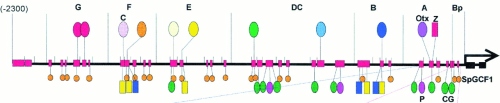
Can the complexity of molecular networks account for the bewildering diversity, e.g., of higher animals and their rapid adaptability to different environments? Interactions between genes are themselves encoded in the genome, and this proves crucial for their dynamics. Specific proteins (called transcription factors) bind to their corresponding target sites on the DNA, thereby enhancing or reducing the activity of the nearest gene. Gene activity has fitness consequences, which in turn adapts the sequences of the target sites on evolutionary time scales through mutations and Darwinian selection. We have analyzed exemplaric cases of this evolution and arrive at a dynamical picture of the genome with relatively static genes ("hardware") and adaptively evolving interactions between them ("software") [1]. We derive consequences for the architecture of genomes in their regulatory regions that compare favorably with genomic data and lead to quantitative predictions of promoter evolution [2].
| [1] | Adaptive evolution of transcription factor binding sites, J. Berg, S. Willmann, and M. Lässig, BMC Evolutionary Biology 4, 42 (2004). |
| [2] | Evolutionary population genetics of promoters: Predicting binding sites and functional phylogenies, V. Mustonen and M. Lässig, Proc. Natl. Acad. Sci. 102, 15936 (2005). |
| [3] | Review article: From biophysics to evolutionary genetics: Statistical aspects of gene regulation, M. Lässig, to appear in BMC Bioinformatics (2007). |
Time-dependent fitness and adaptation
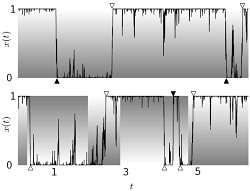
Time-dependent selection causes the adaptive evolution of new phenotypes, and this dynamics can be traced in genomic data. We have developed a new Bayesian inference method for adaptations, which is based on a model for evolution under time-dependent selection and is more sensitive than the standard population-genetic tests [1,2]. We find evidence Analyzing substitutions and polymorphisms in Drosophila by this method shows that selection itself is strongly time-dependent, resembling more a seascape than a static fitness landscape. Our results suggest that selection acts not only as a constraint but as a major driving force of genomic change [1]. The joint statistics of genetic drift and selection fluctuations resembles the physics of systems with quenched disorder [3].
| [1] | Adaptations to fluctuating selection in Drosophila [+Supplementary Information], V. Mustonen and M. Lässig, Proc. Natl. Acad. Sci. 104, 2277-2282 (2007). |
| [2] | A web-based inference tool will be available soon. In the meantime, please contact us, if you are interested in applying this method to your data. |
| [3] | Darwinian evolution under quenched selection, V. Mustonen and M. Lässig, submitted |
Statistical topology of biological networks
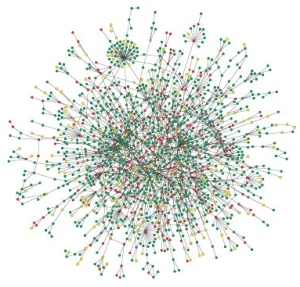
On a coarse-grained level, the statistics of bio-molecular networks can be studied at the level of the network topology (i.e., the ensemble of its nodes and links). Are there topological observables that quantify the deviation of a biological network from a simple random graph? We have established the statistical mechanics of graphs with neighbor connectivity correlations [1]. We have constucted an empirically grounded evolution model for protein networks, which is based on gene duplications and functional mutations of existing proteins and leads to correlated graphs whose structure is in good agreement with the experimental data in yeast [2].
A major application of statistical mechanics to biological networks is in graph alignment. We have established algorithms that find and quantify mutually similar subgraphs within one network or the amount of similarity between different networks, pointing at putative biologically functional units. The underlying statistical theory is grounded on a stochastic evolution model for network topologies. Initial applications have been the identification of functional motifs in the transcription network of E. coli [3] and the analysis of conservation and innovation in gene co-expression networks between human and mouse [4].
| [1] | Correlated random networks, J. Berg and M. Lässig, Phys. Rev. Lett. 89, 228701 (2002). |
| [2] | Structure and evolution of protein interaction networks - A statistical model for link dynamics and gene duplications, J. Berg, M. Lässig, and A. Wagner, BMC Evolutionary Biology 4, 51 (2004). |
| [3] | Local graph alignment and motif search in biological networks, J. Berg and M. Lässig, Proc. Natl. Acad. Sci. 101, 14689 (2004). |
| [4] | Cross-species analysis of biological networks by Bayesian alignment, J. Berg and M. Lässig, Proc. Natl. Acad. Sci. 103, 10967 (2006). |
| [5] | Review article: Bayesian analysis of biological networks: Clusters, motifs, cross-species analysis, J. Berg and M. Lässig, to appear (2007). |
Long-range correlations in the genome
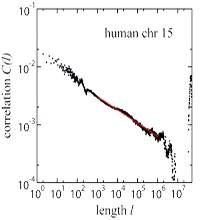
We study a minimal model for genome evolution whose elementary processes are single site mutation, duplication and deletion of sequence regions and insertion of random segments. These processes are found to generate long-range correlations in the composition of letters as long as the sequence length is growing, i.e., the combined rates of duplications and insertions are higher than the deletion rate. For constant sequence length, on the other hand, all initial correlations decay exponentially. These results are obtained analytically and by simulations. They are compared with the long-range correlations observed in genomic DNA, and the implications for genome evolution are discussed [1]. A more comprehensive analyisis of the underlying nonequilibrium stochastic process is contained in [2].
| [1] | A Solvable Sequence Evolution Model and Genomic Correlations, P.W. Messer, P.F. Arndt, and M. Lässig, Phys. Rev. Lett. 94, 138103 (2005). |
| [2] | Universality of long-range correlations in expansion-randomization systems, P.W. Messer, M. Lässig, and P.F. Arndt, J. Stat. Mech. (2005), P10004. |
RNA secondary structures
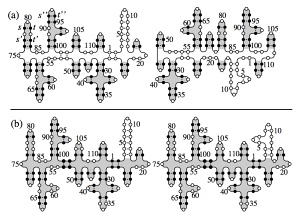
We study secondary structures of random RNA molecules by means of a renormalized field theory based on an expansion in the sequence disorder. We show that there is a continuous phase transition from a molten phase at higher temperatures to a low-temperature glass phase . Primary freezing occurs above the critical temperature: local islands of stable folds form within the molten phase, the size of these islands defining the correlation length of the transition. Our results include critical exponents at the transition and in the glass phase.
| [1] | The freezing of random RNA, M. Lässig and K.J. Wiese, Phys. Rev. Lett. 96, 228101 (2006). |
Statistical theory of alignment
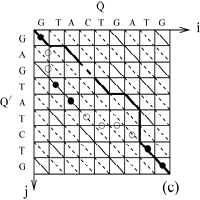
Sequences are similar due to their common evolutionary ancestry, and they differ due to mutations. The recognition and analysis of sequence similarities is one of the most widely used mathematical tools in modern molecular biology. It is crucial to the understanding of genome data. Using concepts and methods from theoretical physics, we have developed a statistical theory of sequence similarity recognition. A recent outcome has been a new algorithm for the detection of distant evolutionary relationships.
| [1] | Similarity detection and localization, T. Hwa and M. Lässig, Phys. Rev. Lett. 76, 2591 (1996). |
| [2] | Scaling laws and similarity detection in sequence alignment with gaps, D. Drasdo, T. Hwa and M. Lässig, J. Comput. Biol. 7, 115 (2000). |
| [3] | Finite-temperature sequence alignment, M. Kschischo and M. Lässig, Pacific Symposium on Biocomputing 5, (2000). |
| [4] | Toward an Accurate Statistics of Gapped Alignment, M. Kschischo, M. Lässig, and Y. Yu, Bull. Math. Biol. 67, 169 (2005). |
